General workflow
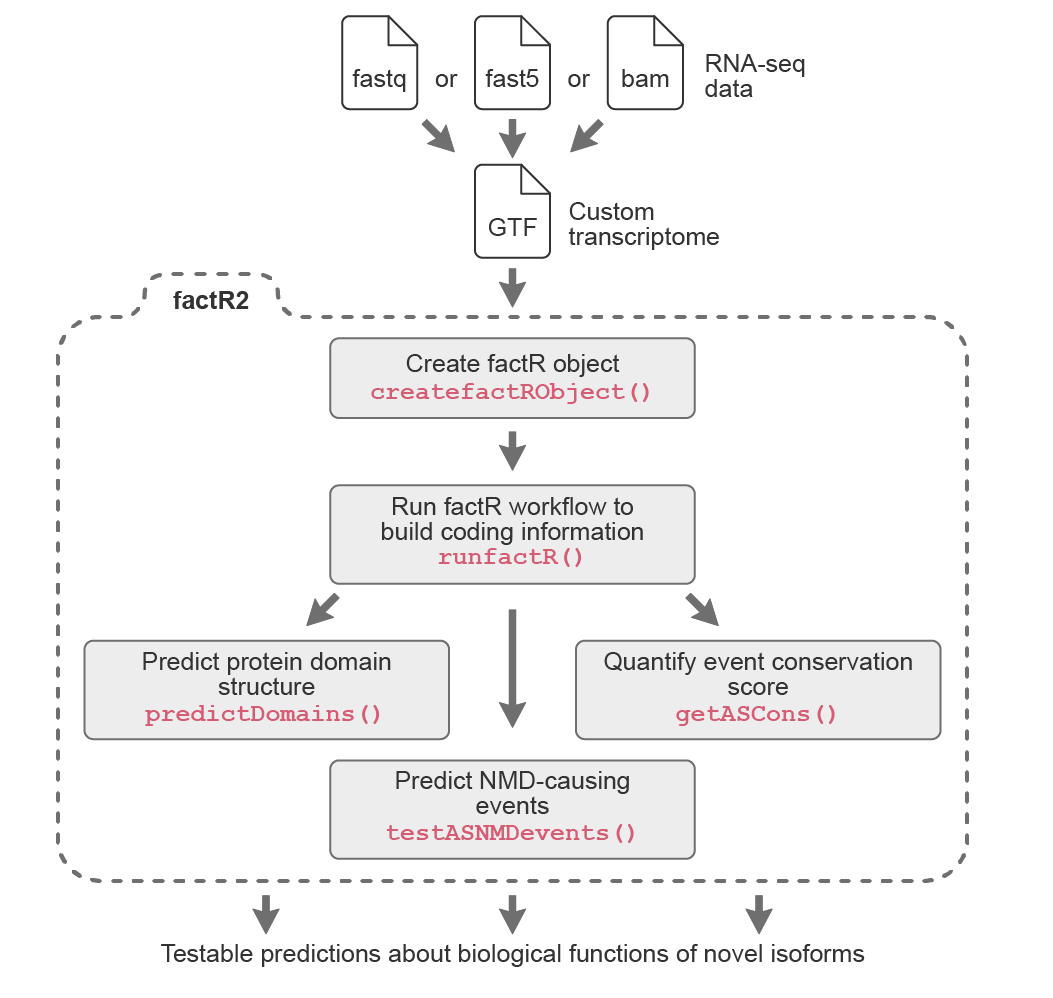
factR2 represents a substantial improvement over factR, providing users with more powerful and user-friendly tools to work with custom-assembled transcriptomes.
Below are factR2’s latest features:
- Extracts alternative splicing events and annotate its novelity and contribution to NMD outcome
- Tests regulatory potential of AS-NMD events
- Quantifies evolutionary conservation scores of alternative exons
- Interactive plot of transcript architectures
As well as the following features from factR:
- Matches gene information on custom transcriptomes to reference annotation
- Constructs transcript coding (CDS) information using reference-guided approach
- Predicts sensitivity of coding transcripts to nonsense-mediated decay (NMD)
- Predicts protein domains on productively spliced transcripts
How to install
The development version can be installed using devtools:
# install.packages("devtools")
devtools::install_github("f-hamidlab/factR2")Getting started
See our full vignette on how to use factR2.